Sequencing Terminology
File Formats
Base calling: A process by which an order of nucleotides in a template is inferred.
FASTA
- A text-based format to store nucleotide or protein sequence
- Part of FASTA software suite developed by Peterson and Lipman in 1985
- First line starts with ">" followed by the identifier
- NCBI defined some standards for the identifiers - Eg:
gi|21434723 - Sequence starts from the second line.
- Each line of sequence is not more than 80 characters
-
Filename extensions
- fasta, fa - Generic
- fna - Nucleic acids
- ffn - Coding region nucleotides
- faa - Amino acids
- frn - Non-coding RNA
- Tools: FASTA, seqkit, DNA Baser, FASTX-toolkit (online tool), Biostrings (R package), etc.
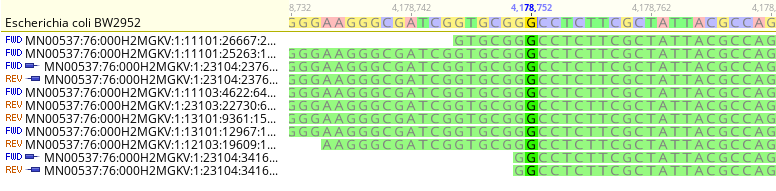
>NR_024570.1 Escherichia coli strain U 5/41 16S ribosomal RNA, partial sequence
AGTTTGATCATGGCTCAGATTGAACGCTGGCGGCAGGCCTAACACATGCAAGTCGAACGGTAACAGGAAG
CAGCTTGCTGCTTTGCTGACGAGTGGCGGACGGGTGAGTAATGTCTGGGAAACTGCCTGATGGAGGGGGA
TAACTACTGGAAACGGTAGCTAATACCGCATAACGTCGCAAGCACAAAGAGGGGGACCTTAGGGCCTCTT
GCCATCGGATGTGCCCAGATGGGATTAGCTAGTAGGTGGGGTAACGGCTCACCTAGGCGACGATCCCTAG
CTGGTCTGAGAGGATGACCAGCAACACTGGAACTGAGACACGGTCCAGACTCCTACGGGAGGCAGCAGTG
GGGAATATTGCACAATGGGCGCAAGCCTGATGCAGCCATGCNGCGTGTATGAAGAAGGCCTTCGGGTTGT
AAAGTACTTTCAGCGGGGAGGAAGGGAGTAAAGTTAATACCTTTGCTCATTGACGTTACCCGCAGAAGAA
GCACCGGCTAACTCCGTGCCAGCAGCCGCGGTAATACGGAGGGTGCAAGCGTTAATCGGAATTACTGGGC
GTAAAGCGCACGCAGGCGGTTTGTTAAGTCAGATGTGAAATCCCCGGGCTCAACCTGGGAACTGCATCTG
ATACTGGCAAGCTTGAGTCTCGTAGAGGGGGGTAGAATTCCAGGTGTAGCGGTGAAATGCGTAGAGATCT
GGAGGAATACCGGTGGCGAAGGCGGCCCCCTGGACGAAGACTGACGCTCAGGTGCGAAAGCGTGGGGAGC
AAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGTCGACTTGGAGGTTGTGCCCTTGAGGCG
TGGCTTCCGGANNTAACGCGTTAAGTCGACCGCCTGGGGAGTACGGCCGCAAGGTTAAAACTCAAATGAA
TTGACGGGGGCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGATGCAACGCGAAGAACCTTACCTGGTC
TTGACATCCACGGAAGTTTTCAGAGATGAGAATGTGCCTTCGGGAACCGTGAGACAGGTGCTGCATGGCT
GTCGTCAGCTCGTGTTGTGAAATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTATCCTTTGTTGCCA...
AB1 format
- Output from Sanger Sequencers (Applied Biosystems)
- Includes quality graphs and electropherograms
- Tools: BioEdit, Chromas Lite, Ugene Pro, MEGA, etc.
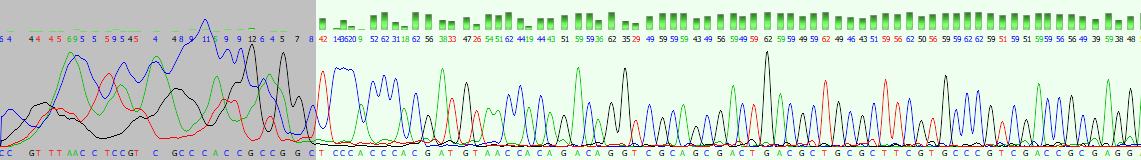
FASTQ
- An extension of FASTA format to store biological data (especially nucleotides)
- It includes quality score for each nucleotide encoded in ASCII characters
- Developed by Sanger Institute
- Default output for sequencers like Illumina
- 1st line - starts with "@" followed by the identifier and optional description
- 2nd line - Sequence itself
- 3rd line - Optional description
- 4th line - Quality scores
- Filename extensions: .fastq, .fq, .fastq.gz, .fq.gz
-
Illumina Sequence Identifiers:
@HWUSI-EAS100R:6:73:941:1973#0/1
@InstrumentID : Flow-cell lane : Tile : Cluster co-x : co-y # Index / Read
@SRR22388518.1 1 length=251
NACTGAAAAACAACAAAAAGCGTTAATCAGTGCGTATAAAAGCGGATTTGACCCTAAAAATGCGGACAAAGTCGCTCAATATTGGCAAAACAAACCCACTAAAATAGACTTACATAAACCTATAAAAACTAAAGACTTCTTTAAAGGGAATACTAATATTTATAGGACACTTCGCAATTTATTTGGACAAAAATTTATGGATAGCTATATTGCTCCTAAAAGTGAAACCACAATGAAAGACTTTATGTCTA
+SRR22388518.1 1 length=251
#<<DDEHIICEHHIHIIIIIIHIHE<GHHIFHHIEHDHHIIHHIIIIIH?1CGHIHIIIIIGEHIIHEHFHIHIIIIDHHIIGHFHIEFEHEFGEGHIIFIGHHHIHIHIIIIIIIIHHIIIIGHIIHHIIIGHHIIHHHHHHHIIICEHHHHHHHIIIFHIHIIECHHHFCEHFHDHHHHFHE?FEHIGIIGHHEGFHGHIEEEHIIIIIBGHHHIHHIFHIFHHH6..6G@HEHHE8F.BFHHHFGHA8
@SRR22388518.2 2 length=251
ATATTCAAGCTATCGGTCCTCATGTAAGTGATCACCCCCATAACGCCTTGTGGGGTGGCTACGCCTTCATATAATTTTTGAGCGATACTCATGGTTTTTGTGGGCGAAAAGCCTAAAAGACTGGAAGCGCTTTGCTGTAAAGTAGAAGTCATGAAAGGGGGCGGTGTGGGGGATTTTTTAGACTTTTTAACGATACTAGAGATAGTGTAGCTTTCTTTTTCCAGTTCGTTTTTAATCTCTTGGGCTTTTTT
+SRR22388518.2 2 length=251
DDDDDIIIIIIIIIIIHIIIIIHIIIIIIIIIIIIIIIIGIIIIIIHIHHIHHHHIIIIIHIIIIIIIHHEHIIIHIHIHIIIHGIIIIIIIIH<D<EHGHGHIHIHHHIIIIHHHHIHIIIIIIHGHHGIIIIHHIIIIGHHIIIHICHH?HHGFCEHHGDHC?CEGIGHHHHE?G?HEFGEHBGGHHECHHHIGHIIIH?GHHH-8@HICHHHIHHII@@@FHIHHBHFHEH@GHIHGG?B@56FEHHH
Phred Quality Score (Q Score)
- Originally developed for Phred program - to automate the calculation of quality scores in the Human Genome Project
- Phred quality scores Q are logarithmically related to the base-calling error probabilities P and defined as Q = -10 log10 P
- For example, if Phred assigns a quality score of 30 to a base, the chances that this base is called incorrectly are 1 in 1000.
| Phred Score | Probability of incorrect base call | Base call accuracy |
|---|---|---|
| 10 | 1 in 10 | 90% |
| 20 | 1 in 100 | 99% |
| 30 | 1 in 1000 | 99.9% |
| 40 | 1 in 10000 | 99.99% |
| 50 | 1 in 100000 | 99.999% |
| 60 | 1 in 1000000 | 99.9999% |
Encoding Q Scores
- Quality scores range from 0-93
- ASCII characters from ! (Decimal value: 33) to ~ (Decimal value 126) are used
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
ASCII
|-------------------------|----|--------|------------------------------|---------------------|
33-----------------------59---64-------73----------------------------104-------------------126
Decimal Value
0........................26...31.......40.....................................................
Sanger
Phred+33
raw reads typically (0, 40)
.........................-5....0........9.............................40......................
Solexa
Solexa+64
raw reads typically (-5, 40)
...............................0........9.............................40......................
Illumina 1.3+
Phred+64
raw reads typically (0, 40)
..................................3.....9..............................41.....................
Illumina 1.5+
Phred+64
raw reads typically (3, 41)
0.2......................26...31........41....................................................
Illumina 1.8+
Phred+33
raw reads typically (0, 41)
0..................20........30........40........50.............65.......................90...
Nanopore
Phred+33
Duplex reads typically (0, 65 + 90)
0..................20........30........40........50.........................................93
PacBio
Phred+33
HiFi reads typically (0, 93)
FAST5 format
- An extension of FAST4 format, which itself is a derivative of FASTQ and part of Swift basecaller
- FAST5 is a type of Hierarchical Data Format 5 (HDF5) used to store large amount of data
- Specifically developed by Oxford Nanopore Technologies
- Guppy - Converts FAST5 files to FASTQ
SAM/BAM
- SAM: Sequence Alignment Map
- Text format for storing sequence data in a series of tab delimited ASCII columns
- Usually it is an output from aligning software (FASTQ reads aligned over Reference Genome)
- Modifications of this format being used to store raw read data - PacBio output format
- Optional Header section - information about the entire file and additional information for alignments
- Alignment section - information for each sequence about where/how it aligns to the reference genome
-
11 Mandatory Columns
Col Field Type Description 1 QNAME String Query template NAME 2 FLAG Int bitwise FLAG 3 RNAME String References sequence NAME 4 POS Int 1- based leftmost mapping POSition 5 MAPQ Int MAPping Quality 6 CIGAR String CIGAR string 7 RNEXT String Ref. name of the mate/next read 8 PNEXT Int Position of the mate/next read 9 TLEN Int observed Template LENgth 10 SEQ String segment SEQuence 11 QUAL String Phred+33 - Can have additional optional columns
- Tools: samtools
BAM: Binary Alignment Map file
Header Section (Optional)
@HD VN:1.0 SO:coordinate
@SQ SN:1 LN:249250621 AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta M5:1b22b98cdeb4a9304cb5d48026a85128
@SQ SN:2 LN:243199373 AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta M5:a0d9851da00400dec1098a9255ac712e
@SQ SN:3 LN:198022430 AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta M5:fdfd811849cc2fadebc929bb925902e5
@RG ID:UM0098:1 PL:ILLUMINA PU:HWUSI-EAS1707-615LHAAXX-L001 LB:80 DT:2010-05-05T20:00:00-0400 SM:SD37743 CN:UMCORE
@RG ID:UM0098:2 PL:ILLUMINA PU:HWUSI-EAS1707-615LHAAXX-L002 LB:80 DT:2010-05-05T20:00:00-0400 SM:SD37743 CN:UMCORE
@PG ID:bwa VN:0.5.4
@PG ID:GATK TableRecalibration VN:1.0.3471 CL:Covariates=[ReadGroupCovariate, QualityScoreCovariate, CycleCovariate, DinucCovariate, TileCovariate], default_read_group=null, default_platform=null, force_read_group=null, force_platform=null, solid_recal_mode=SET_Q_ZERO, window_size_nqs=5, homopolymer_nback=7, exception_if_no_tile=false, ignore_nocall_colorspace=false, pQ=5, maxQ=40, smoothing=1
Alignment Section
1:497:R:-272+13M17D24M 113 1 497 37 37M 15 100338662 0 CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>>
19:20389:F:275+18M2D19M 99 1 17644 0 37M = 17919 314 TATGACTGCTAATAATACCTACACATGTTAGAACCAT >>>>>>>>>>>>>>>>>>>><<>>><<>>4::>>:<9
19:20389:F:275+18M2D19M 147 1 17919 0 18M2D19M = 17644 -314 GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT ;44999;499<8<8<<<8<<><<<<><7<;<<<>><<
| Field | Alignment 1 | Alignment 2 | Alignment 3 |
|---|---|---|---|
| QNAME | 1:497:R:-272+13M17D24M | 19:20389:F:275+18M2D19M | 19:20389:F:275+18M2D19M |
| FLAG | 113 | 99 | 147 |
| RNAME | 1 | 1 | 1 |
| POS | 497 | 17644 | 17919 |
| MAPQ | 37 | 0 | 0 |
| CIGAR | 37M | 37M | 18M2D19M |
| MRNM/RNEXT | 15 | = | = |
| MPOS/PNEXT | 100338662 | 17919 | 17644 |
| ISIZE/TLEN | 0 | 314 | |
| SEQ | CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG | TATGACTGCTAATAATACCTACACATGTTAGAACCAT | GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT |
| QUAL | 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>> | >>>>>>>>>>>>>>>>>>>><<>>><<>>4::>>:<9 | ;44999;499<8<8<<<8<<><<<<><7<;<<<>><< |
GenBank (.gbk .gb)
- Allows storage of meta-information in addition to the sequence
- Human Readable
-
3 main sections
- Header - Starts with LOCUS line
- Features - Coding sequences and their information
- Origin - Sequence
GFF3
- General File Format version 3
- Nine-column, tab-delimited, plain text files
- seqid source type start end score strand phase attributes
- May contain additional lines starting with #
Terminology
| Read | A sequence of nucleotides obtained by sequencing a fragmented genome |
|---|---|
| Insert Size |
The actual length of DNA that is inserted between the adapters Read1+inner_distance(if any)+Read2 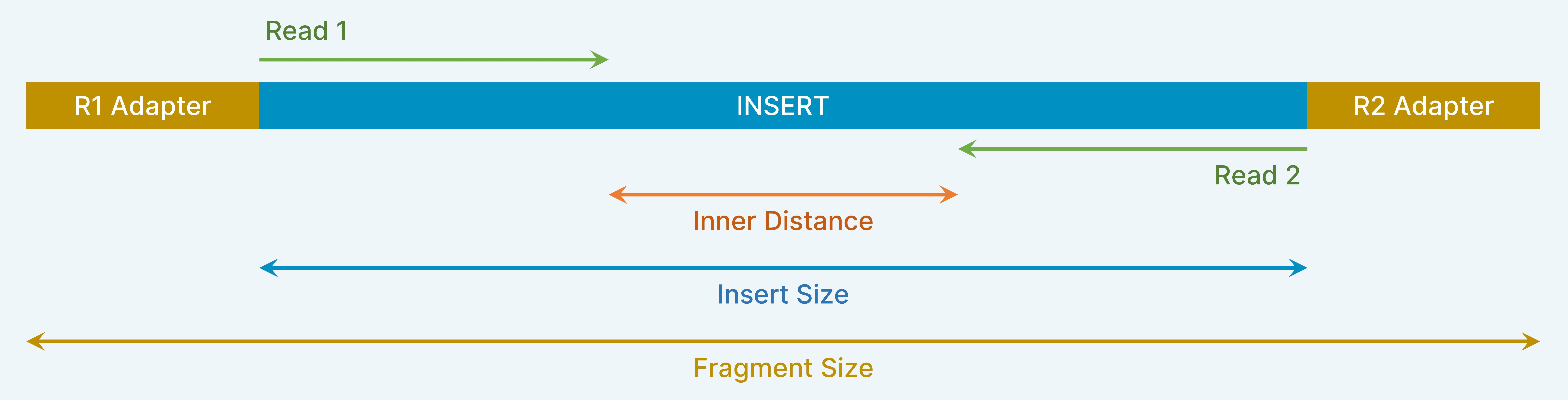
|
| Mate-pairs |
Reads constructed from DNA fragments with longer insert sizes (2-20 kb) Illumina mate-pairs are constructed from 3-5kb DNA fragments |
| Contigs & Scaffolds |
Contiguous sequence: When two sequences overlap at their ends (known as a "dove-tail" overlap), these sequences can be collapsed into a single, non-redundant sequence Scaffolds or supercontig: A scaffold is formed when an association can be made between two contigs that have no sequence overlap 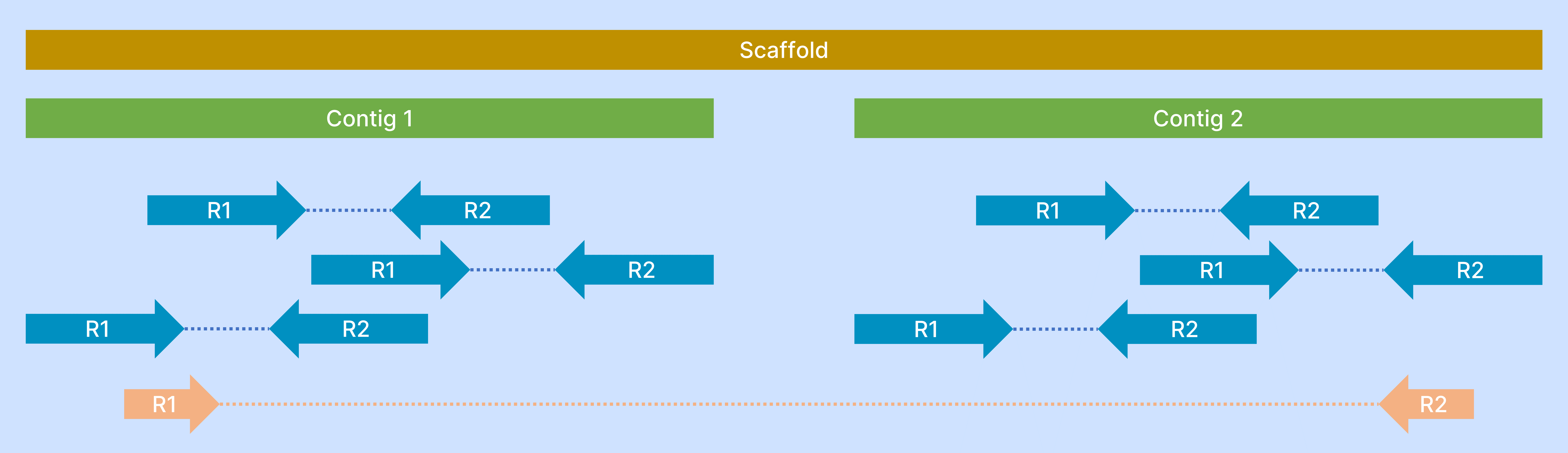
|
| Sequencing Coverage |
The average number of reads that align to, or "cover," known reference bases The Lander/Waterman equation is a method for computing genome coverage. The general equation is: C = LN / G, where C is Coverage, L is Read Length, N is Number of Reads and G is Length of the Genome. |
| N50 |
The shortest contig/scaffold length, at which 50% of the bases in that assembly reside in it and other larger contigs If an assembly has N50 value of 0.8 Mb, this means 50% of the assembled bases are present in contigs/scaffolds of length 0.8 Mb and above Eg: If we have 9 contigs for an assembly with lengths of 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1 Mb. Total genome length is 5.4 Mb (sum of all contigs). Half of which is 2.7 Mb. So, 3 of the large contigs 1 + 0.9 + 0.8 = 2.7 Mb have 50% of the bases. So, N50 = 0.8 Mb. L50 corresponds to the smallest number of contigs that comprise 50% of the assembly. Here, L50 = 3. |
| Draft Genome | A genome sequence that is not yet finished but is of generally high quality. Usually has more than 90% of high quality bases. May include fragments connected with Ns. |
| Gaps | A region of the genome for which no sequence is currently available. Gaps may occur both within and between genomic scaffolds. |
| Genome Annotation | A multi-level process that includes prediction of protein-coding genes, as well as other functional genome units such as structural RNAs, tRNAs, small RNAs, pseudogenes, control regions, direct and inverted repeats, insertion sequences, transposons and other mobile elements |